02/11/2024
Decifrando proteínas

Ganhadores do Prêmio Nobel de Química de 2024, usando computação e inteligência artificial, conseguiram decodificar incríveis estruturas de proteínas.
No último dia 09 de outubro, foram anunciados os ganhadores do Prêmio Nobel de Química de 2024. Eles são David Baker, Demis Hassabis e John M. Jumper, três pesquisadores que, em resumo, "revelaram os segredos das proteínas, usando computação e inteligência artificial".
Para um melhor entendimento da importância das realizações desses cientistas, vamos nos aprofundar um pouco sobre esses temas. Em primeiro lugar, precisamos saber...
O que são proteínas?
A vida como conhecemos não pode existir sem proteínas. Elas controlam e conduzem todos os processos químicos que, em conjunto, são a base da vida física. Além disso, funcionam como hormônios, substâncias sinalizadoras, anticorpos e blocos de construção de diferentes tecidos, tais como músculos, chifres, penas, etc.
Muitas delas são enzimas, que conduzem as reações químicas da vida com precisão surpreendente. Também são importantes as proteínas que ficam nas superfícies celulares, funcionando como canais de comunicação entre as células e seus arredores. A diversidade de seres vivos que conhecemos atesta as incríveis capacidades desses compostos orgânicos como ferramentas químicas.
Geralmente, as proteínas consistem de 20 diferentes aminoácidos, que podem se combinar de infinitas maneiras. Eles podem ser considerados como os "tijolos" de construção da vida.
Usando a informação armazenada no DNA como um projeto, eles se interligam, dentro de nossas células, para formar longas cadeias. Então, a mágica acontece: a sequência de aminoácidos se torce e se dobra, formando uma estrutura tridimensional distinta - às vezes única. Esta estrutura é que dá à proteína a sua função.
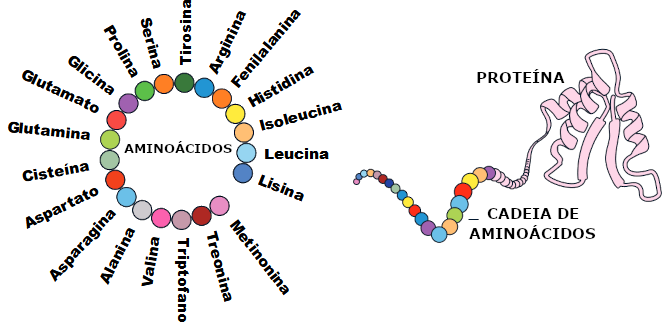
Analisando as sequências de aminoácidos, pesquisadores vinham tentando, desde a década de 1970, prever as estruturas das proteínas. Entretanto, este objetivo sempre se mostrava muito distante e difícil de ser atingido.
Até que, a partir de poucos anos atrás, avanços impressionantes ocorreram, graças aos trabalhos dos...
Nobéis de Química 2024
Metade do prêmio foi para a dupla Demis Hassabis e John Jumper, e a outra metade, para David Baker.
Os dois primeiros apresentaram, em 2020, um modelo de inteligência artificial chamado AlphaFold2 e, com ele, conseguiram desvendar, virtualmente, as estruturas de quase todas as proteínas conhecidas, que são cerca de 200 milhões!
Quanto a Baker, ele teve sucesso na quase impossível missão de dominar os blocos da vida (aminoácidos) e criar proteínas inteiramente novas a partir deles. A primeira delas foi em 2003 e, a partir de então, ele e sua equipe continuaram criando, mais e mais, uma após a outra, inclusive proteínas que podem ser usadas como produtos farmacêuticos, vacinas, nanomateriais e pequenos sensores.
O AlphaFold2 já foi usado por mais de dois milhões de estudiosos, de 190 países. Graças ao modelo de Hassabis e Jumper, os cientistas podem, agora, entender melhor a resistência dos antibióticos e criar imagens de enzimas capazes de decompor o plástico.
A possibilidade de criação de mais e mais proteínas, aliada ao entendimento pleno das estruturas já existentes, representa um potencial científico sem precedentes. São duas vertentes diferentes, porém intimamente relacionadas.
Para conhecer os desafios que os laureados superaram, voltemos ao passado, no alvorecer da Bioquímica Moderna. Continue lendo...
David Baker

Demis Hassabis

John M. Jumper

As primeiras imagens granuladas de proteínas
Os químicos já sabiam, desde o século XIX, o quanto as proteínas são importantes para os processos da vida. No entanto, tiveram que esperar até os anos 1950 para disporem de ferramentas químicas precisas o suficiente para o início de um estudo mais detalhado dessas macromoléculas.
Dois pesquisadores de Cambridge, John Kendrew e Max Perutz, receberam o Prêmio Nobel de Química de 1962, devido a uma descoberta inovadora: eles usaram um método chamado de cristalografia de raios X para obter os primeiros modelos tridimensionais de proteínas.
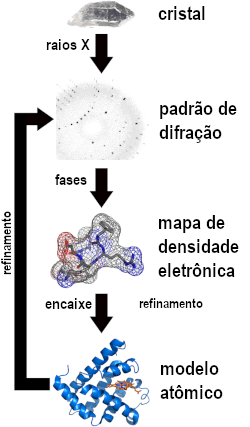
Posteriormente, outros cientistas, usando principalmente o mesmo método (e muito esforço), produziram com sucesso imagens de cerca de 200.000 proteínas diferentes, o que lançou as bases para os trabalhos dos Nobéis de 2024.
Um enigma: como a proteína encontra sua estrutura única?
Em 1972, o químico norte-americano Christian Anfinsen ganhou o Nobel de Química, por ter realizado outra importante descoberta.
Usando vários truques químicos, ele conseguiu fazer com que uma proteína existente se desdobrasse e se dobrasse novamente. O interessante é que a proteína assumiu exatamente o mesmo formato, todas as vezes que o experimento foi repetido.
Em 1961, ele concluiu que a estrutura tridimensional de uma proteína é inteiramente determinada por sua sequência de aminoácidos.
No entanto, a lógica de Anfinsen contém um paradoxo, apontado pelo biólogo molecular Cyrus Levinthal, em 1969. Este cientista, também americano, calculou que, mesmo que uma proteína consista de apenas 100 aminoácidos, teoricamente ela pode assumir uma dentre 1047 estruturas diferentes.
Desta forma, se uma cadeia de aminoácidos for dobrada aleatoriamente, pode levar mais tempo do que a idade do Universo para que a estrutura correta da proteína seja encontrada. Porém, dentro de uma célula, o processo leva apenas alguns milissegundos! Como a cadeia consegue fazer isso? Eis o grande mistério a ser desvendado.
A descoberta de Anfinsen e o paradoxo de Levinthal levam à conclusão de que se trata de um processo predeterminado. Toda a informação sobre como a proteína deve se configurar tridimensionalmente precisa estar presente na sequência de aminoácidos.
O grande desafio da Bioquímica
Tais percepções conduziram a um outro insight decisivo: se a cadeia de aminoácidos é conhecida, tem que ser possível a previsão da estrutura da proteína. Isto significa descobrir exatamente a estrutura tridimensional gerada por determinada sequência.
Esta foi uma ideia excitante. Se os químicos fossem bem-sucedidos em sua implementação, não precisariam mais usar a complicada cristalografia de raios X, economizando bastante tempo, e também seriam capazes de gerar estruturas para todas as proteínas para as quais a cristalografia não fosse aplicável.
Essas conclusões lógicas lançaram o que se tornou o grande desafio da Bioquímica: o problema da predição. No intuito de incentivar um desenvolvimento mais rápido na área, pesquisadores deram início, em 1994, a um projeto chamado Avaliação Crítica da Predição da Estrutura de Proteínas (CASP - Critical Assessment of Protein Structure Prediction).
O projeto foi implementado como uma competição. A cada dois anos, pesquisadores do mundo inteiro tinham acesso a sequências de aminoácidos de proteínas cujas estruturas tinham acabado de ser determinadas, mas permaneciam em segredo para os participantes. O desafio consistia em conseguir prever as estruturas de proteínas, com base nas sequências de aminoácidos conhecidas.
O CASP atraiu muitos pesquisadores, mas a solução do problema se mostrou incrivelmente difícil. As previsões apresentadas pelos participantes não exibiam uma correspondência razoável com as estruturas reais.
De fato, o avanço ocorreu somente em 2018, quando um mestre de xadrez, especialista em Neurociência e pioneiro em inteligência artificial, entrou em campo.
Mestre enxadrista entra nas Olimpíadas da Proteína
Demis Hassabis começou a jogar xadrez aos quatro anos e alcançou o nível de mestre aos treze. Na adolescência, ele iniciou uma carreira de programador e desenvolvedor de jogos de sucesso.
Ele começou a explorar a inteligência artificial (IA) e adotou a Neurociência, área na qual fez várias descobertas revolucionárias. Usando o que aprendeu sobre o cérebro, desenvolveu redes neurais mais eficazes para IA (para saber mais sobre as redes neurais, leia nossa matéria sobre os Nobéis em Física 2024).
Em 2010, foi cofundador da DeepMind, que desenvolveu modelos magistrais de IA para jogos de tabuleiro populares. A empresa foi vendida para o Google em 2014 e, dois anos depois, ganhou projeção global quando conseguiu aquilo que muitos acreditavam ser o Santo Graal da IA: derrotar o campeão de um dos jogos mais antigos do mundo, o Go.
Mas o objetivo de Hassabis não era o jogo. Este era apenas um meio para o desenvolvimento de modelos melhores de IA. Após a vitória no Go, sua equipe estava pronta para enfrentar problemas de maior relevância para a humanidade. Então, em 2018, ele se inscreveu na décima terceira competição CASP.
O modelo IA de Hassabis: uma vitória inesperada
Em anos anteriores, as estruturas de proteínas previstas pelos participantes do CASP tinham atingido uma acurácia de 40 por cento, no máximo. Com seu modelo de IA, o AlphaFold, a equipe de Hassabis chegou a quase 60 por cento.
Foi um excelente resultado, um progresso inesperado que deixou muita gente surpresa, mas a solução ainda não era suficiente. Para ser considerada um verdadeiro sucesso, a previsão precisaria ter uma acurácia de pelo menos 90 por cento em relação à estrutura real.
Então, Hassabi e sua equipe continuaram desenvolvendo o AlphaFold. No entanto, por mais que tentassem, o algoritmo nunca atingia seu objetivo final. A dura verdade era que eles tinham chegado a um beco sem saída.
A equipe já estava cansada, quando John Jumper, um funcionário relativamente novo na empresa, apresentou ideias decisivas sobre como o modelo poderia ser melhorado.
John Jumper assume o grande desafio
O fascínio de John Jumper pelos mistérios do Universo foi o que fez com que ele resolvesse começar a estudar Matemática e Física. No entanto, somente em 2008, quando foi contratado por uma empresa que usava supercomputadores para simular proteínas e sua dinâmica, foi que ele percebeu que o conhecimento de Física poderia ajudar a resolver problemas médicos.
Jumper estava imbuído de seu recém-adquirido interesse em proteínas quando, em 2011, iniciou seu doutorado em Física Teórica. Para economizar capacidade computacional (recurso que era escasso na universidade), ele começou a desenvolver métodos mais simples e engenhosos para a simulação da dinâmica das moléculas da vida.
E ele logo se envolveu de corpo e alma com o grande desafio da Bioquímica. Em 2017, havia completado recentemente seu doutorado, quando ouviu rumores de que a empresa Google DeepMind, em grande segredo, havia começado a prever estruturas de proteínas.
Então, candidatou-se a trabalhar para a DeepMind. A experiência adquirida na empresa anterior o capacitou a ter ideias criativas sobre como aperfeiçoar o AlphaFold. Quando a equipe se firmou, Jumper e Hassabis colideraram um trabalho que reformou fundamentalmente o modelo de IA.
Resultados surpreendentes com o AlphaFold2
A nova versão, o AlphaFold2, foi incrementada com o conhecimento pregresso de Jumper sobre proteínas. A equipe também passou a usar a inovação que vem da recente evolução em IA: redes neurais chamadas transformadores.
Essas redes são capazes de encontrar padrões em enormes quantidades de dados de uma maneira mais flexível que as anteriores, e de determinar com eficiência o que deve ser focado para que haja o atingimento de um objetivo específico.
A equipe treinou o AlphaFold2 nas vastas informações contidas nos bancos de dados sobre todas as estruturas de proteínas e sequências de aminoácidos conhecidas, e a nova arquitetura começou a fornecer bons resultados, ainda em tempo para a décima quarta competição CASP.
Em 2020, quando os organizadores da CASP avaliaram os resultados, eles concluíram que o grande desafio de 50 anos estava vencido. Não maioria dos casos, o AlphaFold2 teve uma performance quase tão boa quanto a da cristalografia de raios X, o que foi surpreendente.
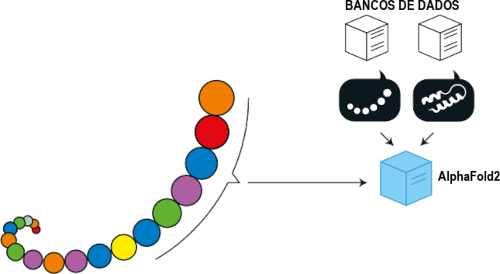
Como o AlphaFold2 funciona?
Com o AlphaFold2 treinado em todas as sequências de aminoácidos e estruturas de proteínas já conhecidas, ele realiza seu trabalho da seguinte forma:
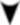
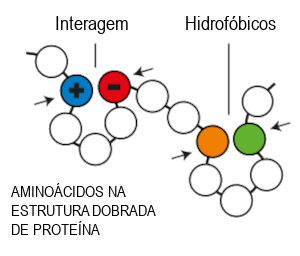
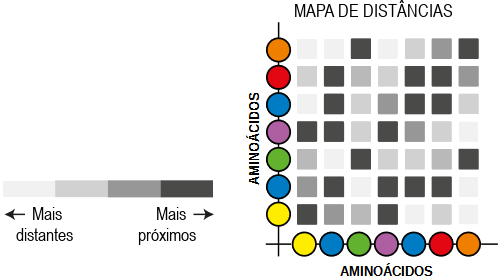
Agora, voltemos um pouco no tempo para destacar outro participante do CASP, ganhador da outra metade do Prêmio Nobel de Química 2024, e sua arte em criar novas proteínas do zero.
A mudança de rumo de David Baker
Quando David Baker ingressou na Universidade de Harvard, ele havia escolhido estudar Filosofia e Ciências Sociais. Entretanto, ao frequentar um curso de Biologia da Evolução, deparou-se com a primeira edição do atualmente clássico livro didático Biologia Molecular da Célula.
Isto fez com que ele mudasse o rumo de sua vida, começando a explorar a Biologia Celular, e foi assim que ficou fascinado pelas estruturas das proteínas.
Quando, em 1993, Baker se tornou um líder de grupo na Universidade de Washington, em Seattle, ele também resolveu encarar o grande desafio da Bioquímica e, usando inteligentes experimentos, começou a explorar como as proteínas se dobram.
Isto lhe trouxe insights que manteve consigo, até que, no final da década de 1990, começou a desenvolver um software que podia prever estruturas de proteínas: o Rosetta.
Baker fez sua estreia na competição CASP em 1998, usando o Rosetta. Em comparação com outros participantes, ele se saiu muito bem, e isto o conduziu a outra ideia: que sua equipe poderia usar o software ao contrário.
Em vez de inserir sequências de aminoácidos conhecidos e obter suas estruturas, eles poderiam ser capazes de inserir uma estrutura desejada e obter sugestões para a sequência, o que lhes permitiria criar novas proteínas!
Baker torna-se um construtor de proteínas
O campo do design de proteínas - onde essas estruturas são criadas com novas funções - começou a decolar no final da década de 1990. Pesquisadores andaram ajustando proteínas já existentes para que elas pudessem fazer coisas como quebrar substâncias perigosas ou funcionar como ferramentas na indústria química.
Mas isto não satisfez David Baker e sua equipe. A gama de proteínas naturais é muito limitada. Por isso, para aumentar o potencial de obtenção de proteínas com novas funções, eles queriam criá-las do zero!
O grupo arquitetou uma proteína com uma estrutura nova, ainda não existente na natureza e, então, pediu a Rosetta para calcular qual sequência de aminoácidos poderia resultar nessa nova proteína.
Rosetta, então, pesquisou um banco de dados de todas as estruturas de proteínas conhecidas e procurou por fragmentos que fossem similares à estrutura desejada. Usando seus conhecimentos sobre o cenário energético das proteínas, Rosetta otimizou esses fragmentos e propôs uma sequência de aminoácidos.
Para investigar se esta sequência foi bem-sucedida, a equipe introduziu, em bactérias, o gen que induziria a produção da proteína em questão. As bactérias produziram quase que exatamente a proteína desejada, então eles conseguiram determinar sua estrutura usando cristalografia de raios X.

Criações espetaculares
David Baker publicou sua descoberta em 2003. A Top7, primeira proteína inteiramente nova e com 93 aminoácidos, era maior do que qualquer coisa do tipo já produzida anteriormente. Na verdade, até então só se conseguia imitar estruturas já existentes.
E este foi apenas um primeiro passo de algo que pode ser descrito como uma evolução extraordinária, pois ele e sua equipe continuaram criando mais e mais novas proteínas, com funções, inclusive, inéditas entre as proteínas criadas pela natureza.
Após a competição CASP de 2020, quando Baker percebeu o potencial dos modelos de IA baseados em transformadores, ele adicionou um ao Rosetta, e isto facilitou ainda mais o design. Veja, na ilustração a seguir, algumas de suas espetaculares criações:
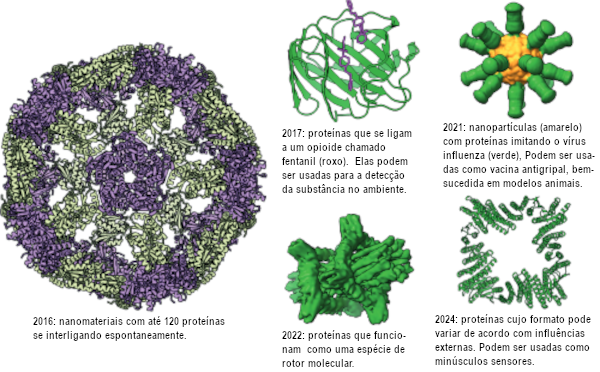
[Imagens: ©Johan Jarnestad/The Royal Swedish Academy of Sciences]
O código-fonte do Rosetta foi disponibilizado, permitindo que uma equipe global de pesquisadores o continuassem desenvolvendo e, assim, descobrissem novas áreas de aplicação.
Um trabalho de anos passou a levar poucos minutos
Quando Demis Hassabis e John Jumper confirmaram que o AlphaFold2 realmente funcionava, eles calcularam a estrutura de todas as proteínas do corpo humano. A partir daí, foram ainda mais longe: previram a estrutura de todas as 200 milhões de proteínas que os pesquisadores conseguiram descobrir nos organismos da Terra, até o momento.
A Google DeepMind também disponibilizou publicamente o código do AlphaFold2, e o modelo tornou-se uma mina de ouro para os pesquisadores. O software já foi usado por mais de 2 milhões de pessoas, de 190 países.
Anteriormente, muitas vezes eram necessários anos para a obtenção da estrutura de uma proteína, isto se o objetivo fosse de fato alcançado. Agora, o processo pode ser realizado em poucos minutos.
O modelo de IA não é perfeito, mas estima a correção da estrutura que produziu. Assim, pode-se saber o quão confiável é o resultado. A figura abaixo mostra alguns dos muitos exemplos de como o AlphaFold2 pode abrir campo para fantásticos avanços:
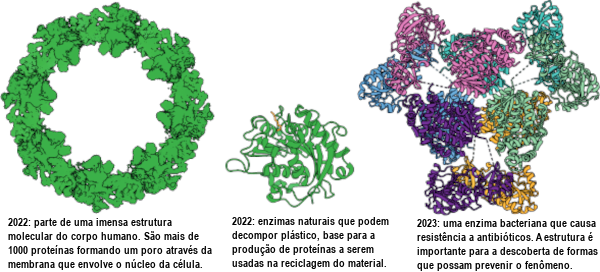
[Imagens: ©Johan Jarnestad/The Royal Swedish Academy of Sciences]
Uma vertiginosa evolução em benefício da humanidade
A incrível versatilidade das proteínas como ferramentas químicas reflete-se na vasta diversidade da vida. O fato de podermos, agora, visualizar com tanta facilidade a estrutura dessas pequenas máquinas moleculares é algo alucinante.
Isto acarreta um melhor entendimento de como a vida funciona, por que algumas doenças se desenvolvem, como ocorre a resistência a antibióticos, por que alguns micróbios podem decompor plástico, etc...
Fica evidente como as descobertas dos três ganhadores do Prêmio Nobel de Química de 2024 trazem consigo um potencial de proporcionar incríveis benefícios para a humanidade!
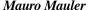 - publicada em 02/11/2024.
- publicada em 02/11/2024.★ A maior parte do conteúdo principal resulta de estudo, tradução e adaptação das seguintes fontes:
- The Nobel Foundation (nobelprize.org);
- Royal Swedish Academy of Sciences - The Nobel Prize in Chemistry 2024: Popular Science Background .




